搜索
关闭 X




复旦大学潘翔城Nat. Synth.:1,2–硼迁移驱动的自由基C3交替聚合
发布时间:2026-03-25
自由基聚合因其操作简便、反应条件温和、和单体范围广而成为聚合物合成的基石。传统自由基聚合在将C2(例如丙烯酸酯、苯乙烯和丙烯酰胺)和C4单体(例如共轭二烯烃)引入聚合物结构方面取得了显著成功,推动了广泛的工业应用。然而,开发一种高效的自由基聚合策略以实现C3结构的引入仍然是聚合物化学中的一个未解决的难题(图1a)。这一限制源于自由基聚合中的机制约束:传统C2自由基聚合通过对乙烯基单体的β-碳(C1)进行区域选择性自由基加成,在α-位置(C2)产生一个稳定的碳自由基。关键是,不存在类似的机制来通过单体加成在γ-位置(C3)产生自由基,也不能稳定这样的C3碳自由基中间体。这一机制约束阻碍了有效的C3结构引入,是自由基聚合中的关键方法学挑战。
复旦大学高分子科学系和聚合物分子工程全国重点实验室潘翔城教授团队长期致力于自由基聚合新方法的发展,尤其是提出了“杂原子自由基可控聚合”(J. Am. Chem. Soc.2025, 3662-3669; 2022, 19942-19952; 2021, 19167-19177; Nat. Commun.2024, 15:6179; 2021, 12:5853; Angew. Chem. Int. Ed.2026, e21319; 2025, e202506972; 2018, 9430-9433),将硼自由基引入聚合体系中。针对上述问题,作者开发了一种基于硼自由基的交替聚合策略,该策略协同结合了1,2-硼迁移与自由基极性匹配来克服C3结构引入的挑战(图1b)。
相关研究成果以题为“Radical C3 alternating polymerization driven by 1,2-boryl migration via radical polarity matching”最近发表于《Nature Synthesis》上。徐超然博士、硕士研究生孔誉为该论文共同第一作者、Michelle L. Coote院士和潘翔城教授为本文共同通讯作者。特别感谢国家自然科学基金、上海市基础研究特区计划、尤其是上海泰坦自然科学发展基金会对该工作的支持。
在设计的机制中,亲电自由基最初攻击C3结构的α-烯烃单体,产生仲碳自由基中间体。这些中间体经历快速1,2-硼迁移,产生更加稳定的三级碳自由基。随后选择性地加成到缺电子的共聚单体上,再生成亲电自由基,建立一个自我维持的极性匹配过程。该方法实现了聚合物序列的精确控制,同时成功地将以前难以引入的C3结构单元纳入到交替共聚物中。
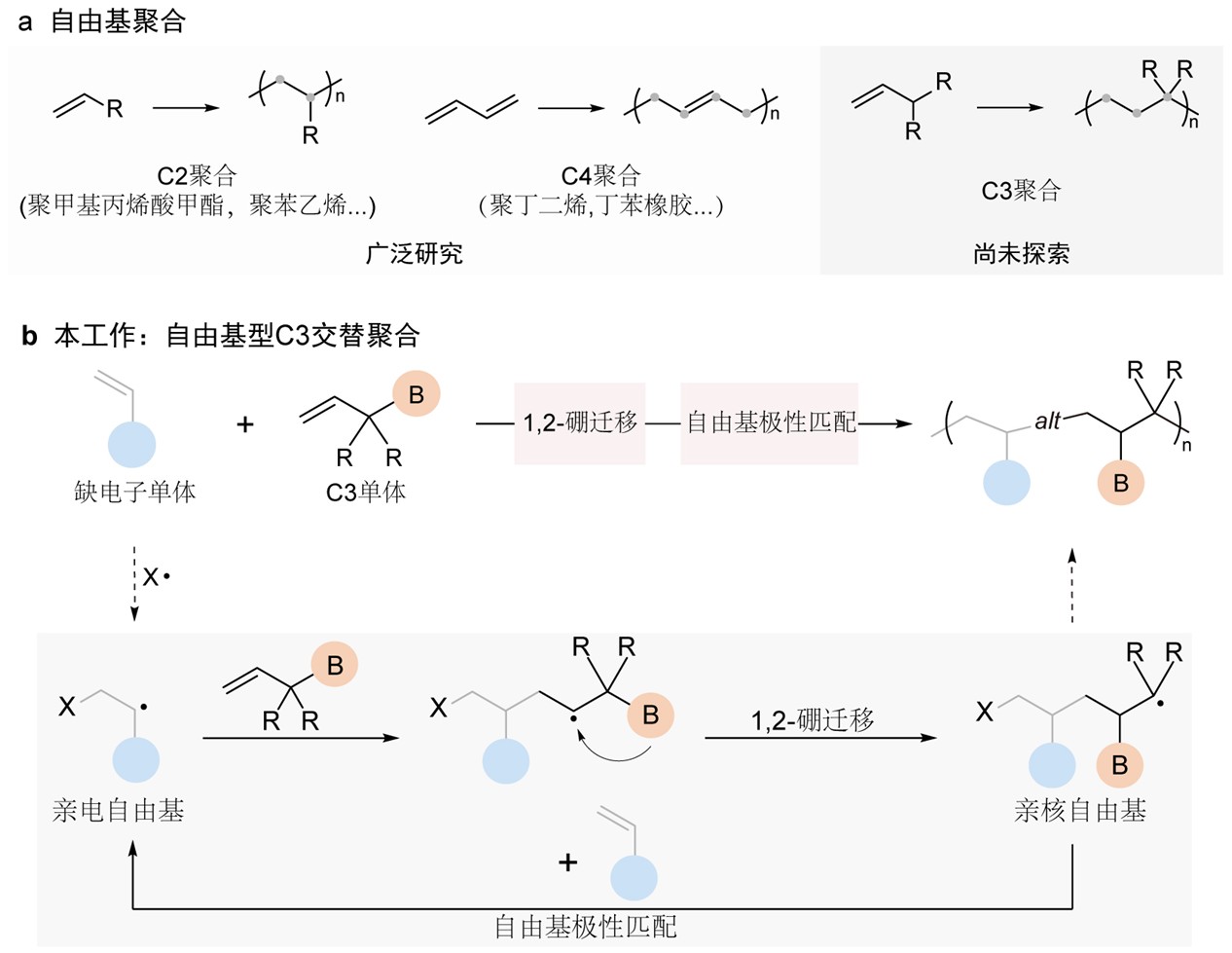
图1. 自由基型C3交替聚合
通过调节聚合反应条件,获得数均分子量为17.2 k,分散度为1.76的交替聚合物。结构表征和聚合反应动力学研究证实了交替聚合反应的进行。作者评估了一系列α-取代基结构不同的烯丙基硼酸酯C3单体,以阐明成功进行交替共聚合所需的结构要求。通过筛选不同的共聚单体,验证了自由基极性匹配原理的重要性。
为了阐明其内在机理,研究团队结合了模型反应与密度泛函理论计算。通过三氟甲基自由基引发M1的模型反应,成功捕获了1,2-硼迁移后的产物,直接验证了该关键步骤的存在。DFT计算进一步揭示了反应的能量路径(图2)。计算表明,对于成功的单体M1,1,2-硼迁移将不稳定的仲碳自由基转化为更稳定的叔碳自由基,该过程在动力学和热力学上均高度有利,其速率远超与单体直接加成。迁移后生成的亲核自由基对缺电子的MAH的加成速率极高,确保了严格的交替序列。
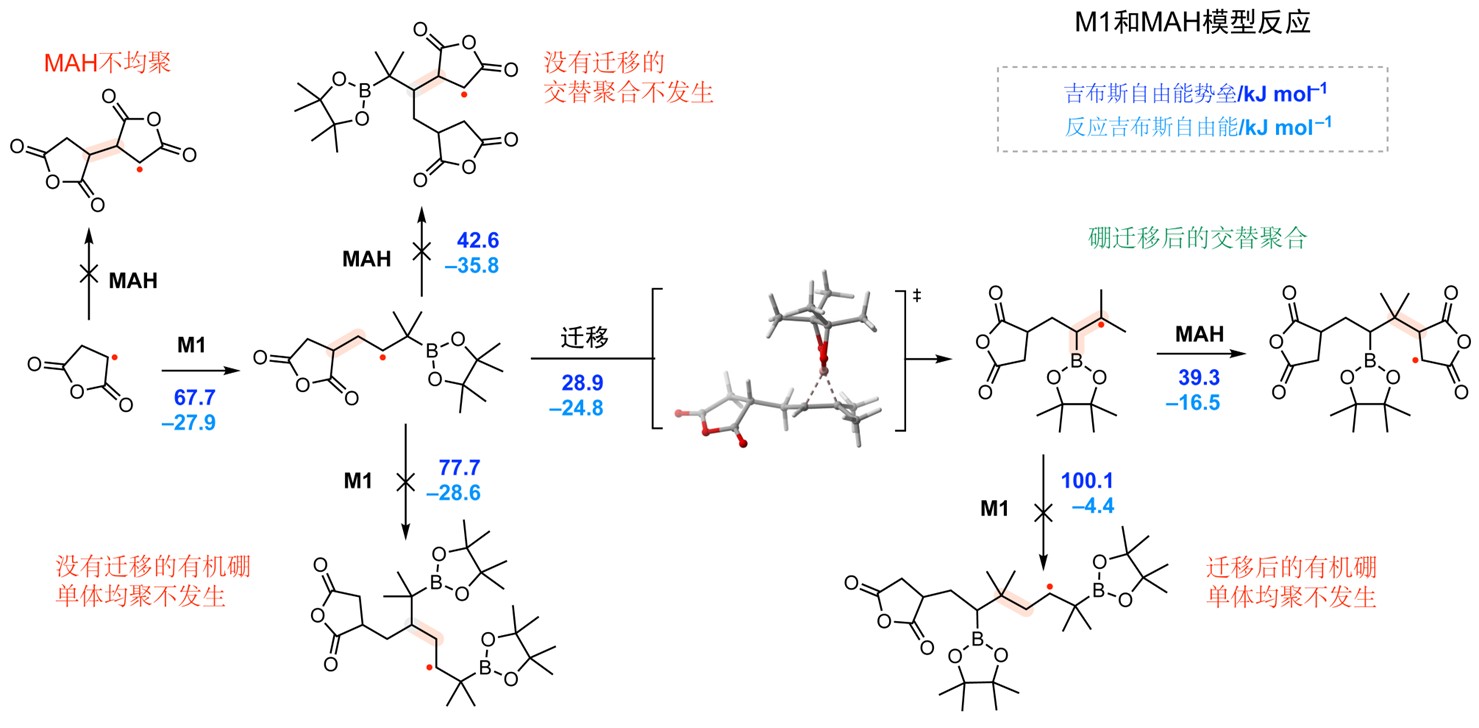
图2. M1单体与MAH自由基C3交替聚合的DFT计算
这项研究工作成功突破了自由基C3聚合的长期瓶颈,通过结合1,2-硼迁移与自由基极性匹配,发展了一种高效的交替共聚策略。该策略不仅为将C3结构单元引入聚合物主链提供了通用平台,还同时将具有高反应活性的硼酸酯官能团引入聚合物,为后续的聚合物后修饰和功能化材料开发打开了新的大门。展望未来,这种自由基迁移驱动的聚合策略有望进一步扩展自由基聚合的单体范围,为合成具有精确序列结构和可编程功能的新型高分子材料提供强大工具。
论文题目:Radical C3 alternating polymerization driven by 1,2-boryl migration via radical polarity matching
全文链接:https://www.nature.com/articles/s44160-026-01005-8
doi: 10.1038/s44160-026-01005-8
课题组网站http://www.panxlab.com(常年招收对高分子合成感兴趣的博士后研究员、硕士和博士研究生等)

复旦高材生
微信公众平台
订阅号:FDUMMers
Copyright© 2012 复旦大学高分子科学系 邮编 :200438 传真:021-31242888 沪ICP备05003394
技术支持: 维程互联